 Login/Register
Login/Register 




TotalSeq™-A Antibodies and Cell Hashing with 10x Single Cell 3' Reagent Kit v3.1 (Dual Index) Protocol
The following protocol describes surface protein staining with TotalSeq-A antibodies and/or hashtag antibodies, to enable protein detection with 10x Genomics Chromium Single Cell 3' Reagent Kits. This protocol has been optimized using liquid, single-antibody TotalSeq-A conjugates, and TotalSeq-A cocktails.
Read through this protocol in its entirety and the associated 10x Genomics user guide you intend to use prior to starting so you can plan accordingly. TotalSeq-A antibodies and cocktails are compatible with the following 10x Genomics 3’ assays and user guides.
For customers using TotalSeq™-B or TotalSeq™-C antibodies, please refer to our TotalSeq™-B or C protocol.
Note:
User is solely responsible for determining whether there are any licenses or other intellectual property rights necessary for user’s intended use of any BioLegend products or any protocol or with any other products or technologies and assumes all risk and liability arising from such use.
Commonly Used Abbreviations and Terminology:
- ADT – Antibody Derived Tag. Refers to a TotalSeq DNA-barcoded oligonucleotide that is directly conjugated to a specific antibody clone of interest. Each antibody clone was raised against a specific extracellular protein epitope and can be used to characterize the expression of that surface antigen on cells. Thus, ADTs serve as DNA tags used to catalog and quantify distinct surface protein expression levels.
- HTO – Hash Tag Oligonucleotide. Refers to a TotalSeq DNA-Barcoded Oligonucleotide that is directly conjugated to a cocktail of two independent antibody clones that are specific for surface proteins known to be ubiquitously expressed on various cell types. The intention of an HTO is to enable researchers to load multiple samples into a single reaction and maintain the ability to determine sample origin.
- CITE-Seq – Cellular Indexing of the Transcriptome and Epitopes by Sequencing. This application was first described by Stoeckius et al. (Nat Methods 14, 865–868 (2017)) of the NY Genome Center as they used antibodies coupled with oligonucleotides to simultaneously measure proteins and RNA at a single-cell level.
- Cell Hashing – Application where oligo-tagged antibodies against ubiquitously expressed surface proteins uniquely label cells from distinct samples, which can be subsequently pooled. By sequencing these tags alongside the cellular transcriptome, users can assign each cell to its original sample.
Explore our TotalSeq Resources and Learning webpage to discover technical resources and publications that showcase the utilization of TotalSeq-A antibodies.
Protocol Overview
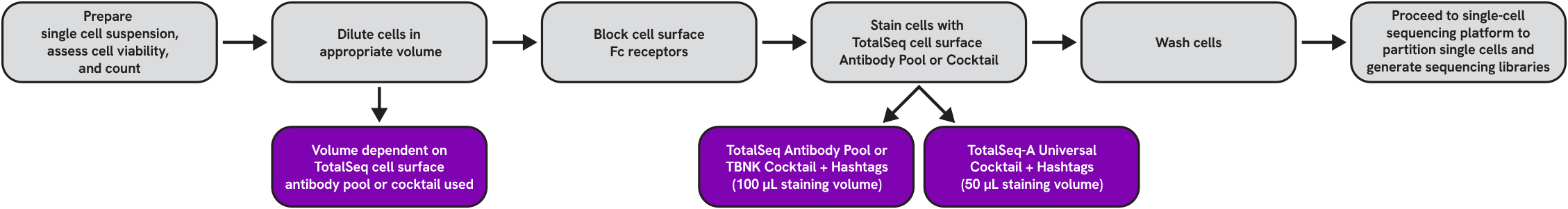
Reagent and Consumable List
- Cell Staining Buffer (BioLegend, Cat. No. 420201)
- Human TruStain FcX™ (Fc Receptor Blocking Solution) (BioLegend, Cat. No. 422301)
- TruStain FcX™ PLUS (anti-mouse CD16/32) (BioLegend, Cat. No. 156603)
- Low Protein Binding Microcentrifuge Tubes (ThermoFisher Scientific, Cat. No. 90410 or equivalent)
- 12 x 75 mm Falcon™ Round-Bottom Polystyrene Tubes (Fisher Scientific, Cat. No. 14-959-1A or equivalent)
- Nuclease-free Water (Thermo Fisher, Cat#AM9937 or equivalent)
- Ethanol (Sigma, Cat# E7023-500ML or equivalent)
- Nuclease-Free Pipette Tips (e.g. Thermo Fisher Scientific AM12650, AM12660, or equivalent)
- TempAssure PCR 8-strips (USA Scientific, Cat# 1402-4700 or equivalent)
- PCR Thermocycler (Bio-Rad, T100™ Thermal Cycler or equivalent)
Optional
- Curiox Laminar Washing- The Curiox Laminar Wash system can be used to wash cells stained with TotalSeq antibodies. For guidance on use, please contact Curiox or BioLegend Technical Services.
Reagents for Library Preparation
Important: cDNA additive and dual Index primers must be ordered from 3rd party vendors prior to starting the protocol.
- Quantabio sparQ HiFi PCR Master Mix (2X) (Quantabio, Cat# 95192-250) or KAPA HiFi HotStart ReadyMix (2X) (Kapa Biosystems, Cat# KK2601)
- Quantabio sparQ PureMag Beads (Quantabio, Cat# 95196-005) or SPRIselect reagent (Beckman Coulter, Cat# B23317)
- 4200 TapeStation (Agilent Technologies, Cat# G2991A)
- DNA High Sensitivity D1000 and High Sensitivity D5000 (Agilent, Cat# 5067-5584/5067-5592)
- Qubit™ 3 (Thermo Fisher Scientific, Cat# Q33226)
- Qubit™ dsDNA HS Assay Kit (Thermo Fisher, Cat# Q32854/Q32851)
- cDNA amplification primers: ADT additive primer (0.2µM Stock) and/or HTO additive primer v2 (0.2 µM Stock)
- ADT and/or HTO Library Construction primers: Dual Index ADT i5/i7 Primer Pairs (DI_ADTx) (10µM Stock) and/or Dual Index HTO i5/i7 Primer Pairs (DI_HTOx) (10 µM Stock)
See notes at the end of the protocol for further details on primer sequences. For a full list of sequences, view the primer sequence table below or download this table. Please contact Technical Services if you have questions.
Additional Suggested Reagents
- TotalSeq™-A Antibodies
- TotalSeq™-A Human Universal Cocktail, V1.0, (BioLegend, Cat No. 399907)
- TotalSeq™-A Human TBNK Cocktail, (BioLegend, Cat. No. 399901)
- TotalSeq™-A Mouse Universal Cocktail, V1.0, (BioLegend, Cat. No. 199901)
Best Practices and Important Considerations for Best Results
Surface Staining
Cell washing
- When washing cells, it is extremely important to thoroughly decant the wash buffer, and, upon the addition of new wash buffer, that the cell pellet is resuspended either with pipette mixing or gentle vortexing. When decanting, pour off the wash buffer in a single firm, but not forceful motion. Following decanting, continue to hold the tube inverted and remove the remaining droplets on the lip of the tube by gently dabbing with a clean paper towel before returning the tube to an upright position. This technique should be used during all cell washes.
Optimal cell staining with TotalSeq™ antibodies
- Liquid single-antibody TotalSeq conjugates require optimization of staining concentration (titration) to obtain the best results, as performed by the end user of the antibodies. Optimization of antibody staining concentration is essential to obtaining good quality data in antibody-based applications such as surface staining using TotalSeq antibodies.
- Antibody titration for sequencing-based applications using TotalSeq antibodies is best performed using matching procedures as much as possible. This means titration-by-sequencing if possible. In the absence of titration-by-sequencing, it may be possible to replicate this protocol using fluorescent antibodies of the same clone and titrate via flow cytometry. For more information regarding titration of TotalSeq-A antibodies, please read Tips and Tricks for Titrating TotalSeq Antibodies. BioLegend can provide recommended concentration ranges for most TotalSeq antibodies, and these can be obtained by contacting BioLegend Technical Services.
- Throughout this protocol, the term “antibody pool” is defined as a user-created antibody pool of titrated single TotalSeq antibodies.
- BioLegend’s lyophilized Human TotalSeq-A antibody cocktails have been validated for use with this application. The cocktails are optimized for specific staining reaction sizes. The TotalSeq-A Human TBNK Cocktail is optimized to stain 1 x 106 cells in 100 µL staining volume, and all other TotalSeq-A Cocktails have been optimized to stain 5 x 105 cells in 50 µL staining volume.
Note:
We have demonstrated staining of 2.5 x 105 - 2 x 106 cells from PBMCs with 1 test of our TotalSeq-A Cocktails with no effect on staining efficiency allowing for reliable detection of lineage markers using this protocol. Sample specific variations on cocktail performance are expected with smaller or larger cell input numbers and customers should verify adequate sensitivity in cocktail performance in their experiment. It is important that final staining volume, regardless of cell number used, not exceed 100 µL for the TBNK cocktail and 50 µL for all other TotalSeq-A lyophilized cocktails.
Protocol
I) Cell Surface Staining
- Prepare cell suspension with preferred or recommended method
Notes:
- This protocol has been optimized using fresh human PBMCs isolated using density gradient centrifugation. Whole blood or lysed whole blood is not recommended. If using cells isolated with a different procedure, users may need to verify the antibody staining pattern using alternative methods.
- BioLegend has not tested this protocol using single-cell suspensions derived from enzymatically digested tissue. Enzymatic digestion may result in alterations of surface protein epitopes and impact staining with TotalSeq antibodies. Optimization of staining conditions and concentrations may be required.
- Count and assess cell viability
- Using your preferred method, carefully count all cells to ensure accurate quantitation and assess cell viability.
Note:
Contact BioLegend Technical Services with any questions regarding cell viability. Ideal cell viability is >95%. Low cell viability is associated with poor single-cell sequencing data. If low cell viability is observed, users may need to enrich live cells or repeat cell suspension preparation.
- Using your preferred method, carefully count all cells to ensure accurate quantitation and assess cell viability.
- Dilute cells in an appropriate volume prior to staining
- When using an antibody pool or the TotalSeq-A TBNK Cocktail:
- If working with human cells, dilute 1 x 106 cells in 45 μL of Cell Staining Buffer in a 12 x 75 mm flow cytometry tube.
- If working with mouse cells, dilute 1 x 106 cells in 49.5 μL of Cell Staining Buffer in a 12 x 75 mm flow cytometry tube.
- When using any other TotalSeq-A Cocktail:
- If working with human cells, dilute 5 x 105 cells in 22.5 μL of Cell Staining Buffer in a 12 x 75 mm flow cytometry tube.
- If working with mouse cells, dilute 5 x 105 cells in 24.75 μL of Cell Staining Buffer in a 12 x 75 mm flow cytometry tube.
- When using an antibody pool or the TotalSeq-A TBNK Cocktail:
- Fc receptor blocking
- If using an antibody pool or the TotalSeq-A TBNK Cocktail:
- For human cells, add 5 μL of Human TruStain FcX to 1 x 106 cells in 45 µL of Cell Staining Buffer (total volume = 50 μL).
- For mouse cells, add 0.5 µL of TruStain FcX PLUS (anti-mouse CD16/32) blocking reagent to 1 x 106 cells in 49.5 µL of Cell Staining Buffer (total volume = 50 μL).
- If using any other TotalSeq-A Cocktail:
- For human cells, add 2.5 μL of Human TruStain FcX Fc blocking reagent to 5 x 105 cells in 22.5 µL of Cell Staining Buffer (total volume = 25 μL).
- For mouse cells, add 0.25 μL of TruStain FcX PLUS (anti-mouse CD16/32) blocking reagent to 5 x 105 cells in 24.75 µL of Cell Staining Buffer (total volume = 25 μL).
- Incubate for 10 minutes at 4°C.
- While cells are incubating in Fc Block, proceed to step 5 – Stain cells with cell surface antibodies.
- If using an antibody pool or the TotalSeq-A TBNK Cocktail:
- Stain cells with cell surface antibodies
Notes:
- If you plan to multiplex samples using TotalSeq hashtags, there are two commonly used approaches to staining samples with hashtag antibodies.
- Option 1: Individual samples are stained with TotalSeq antibodies (user-created antibody pool or with our pre-optimized TotalSeq-A cocktails) and hashtags in a single step, washed three times to remove unbound antibodies, and subsequently pooled for loading into one or more single-cell sequencing reactions. See figure 1A.
- Option 2: Involves first staining individual samples with hashtags, washing three times, sample pooling, and then staining with the TotalSeq antibodies (user-created antibody pool or with our pre-optimized TotalSeq-A Cocktails), followed by another three washes. See figure 1B.
- Whenever possible, we recommend option 1, as option 2 can result in increased sample loss and reduced cell viability due to multiple wash steps. If using option 2, we do not advise reducing sample washing as this may result in higher background and poor data quality. Additional information regarding use of TotalSeq hashtag antibodies can be found here – Efficient Multiplexing With TotalSeq™ Hashtags.
- For assistance in de-multiplexing after sequencing, please contact BioLegend Technical Services.
- If cell hashing samples simultaneously with our TotalSeq Cocktails, or if you plan to “Spike-In” additional antibodies to the cocktails, please refer to our TotalSeq Antibody Cocktail “Spike-in” Guidance protocol prior to reconstitution of the lyophilized cocktail to determine how to make the Cell Staining Buffer and Spike-in reconstitution mix.
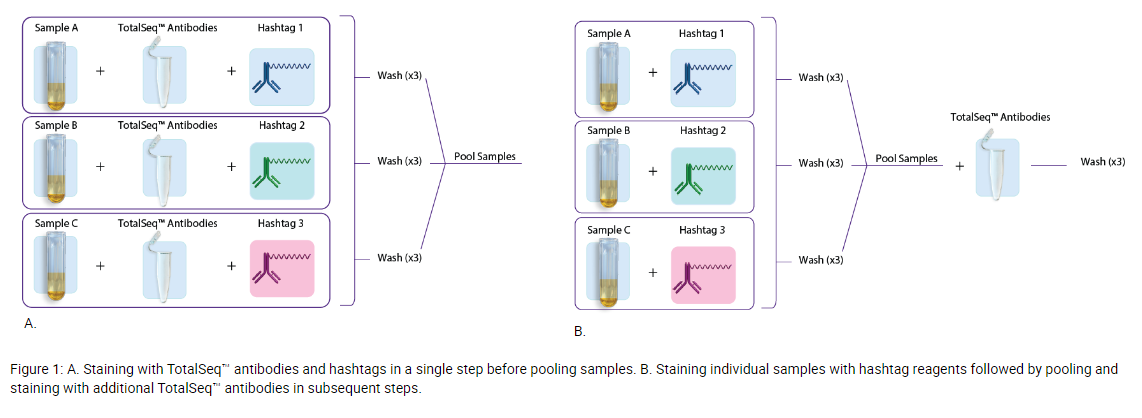
- Staining individual samples with TotalSeq™ Hashtag Antibodies for sample pooling
Notes:
- Proceed to step 5.2 if one of the following applies:
- If you are not using TotalSeq Hashtags.
- If you are staining cells simultaneously with an antibody pool or TotalSeq-A cocktail and hashtags in a single step before pooling samples (figure 1A).
- If sequentially staining individual samples with hashtag antibodies, pooling samples, and then staining with a TotalSeq-A antibody pool or one of our TotalSeq-A Cocktails as seen in figure 1B, follow the steps below before staining your samples with an antibody pool or cocktail. Please note that when sequentially staining, cell recovery may be diminished, and cell viability may be negatively impacted in some cases.
- Make a unique hashtag staining solution for each sample using a titrated amount of each hashtag in Cell Staining Buffer. Add the calculated amount of each hashtag antibody to a low protein binding microcentrifuge tube, and bring up the total volume with Cell Staining buffer to 50 µL if using an antibody pool or TotalSeq-A TBNK cocktail. Or, bring the total volume up to 25 µL if using any other TotalSeq-A Cocktail.
- Centrifuge the hashtag solution at 14,000 x g at 2 – 8°C for 10 minutes before adding to the cells to ensure the removal of protein aggregates.
- Carefully pipette out the prepared hashtag solution, avoiding the bottom of the tube, and add it to the 50 µL or 25 µL blocked cell suspension.
- Incubate for 30 minutes at 4°C.
- Wash cells by adding 3 mL of Cell Staining Buffer, and mix by gently pipetting 5 times. Centrifuge at 4°C for 5 minutes at 400 – 600 x g depending on your sample type.
- Repeat wash twice for a total of 3 washes.
Note:
It is extremely important to thoroughly decant the wash buffer and resuspend the cell pellet either with pipetting or gentle vortexing. Discard supernatant with a single firm, but not forceful motion. Proceed to absorb any remaining liquid on the lip of the tube with a clean paper towel.
- After the final wash, resuspend each sample in 100 µL of Cell Staining Buffer, and verify cell concentration for each sample.
- Based on each sample’s cell concentration, combine an equal number of cells from each sample to achieve 1 x 106 cells if using an antibody pool or the TotalSeq-A TBNK Cocktail, or 5 x 105 cells if using any other TotalSeq-A Cocktail in a low protein binding microcentrifuge tube. The final volume will be dependent on the specific staining reaction size of the antibody pool or cocktail being used.
- If staining cells with an antibody pool or the TotalSeq-A TBNK cocktail and the volume of combined cells is less than 50 µL, adjust volume with Cell Staining Buffer up to 50 µL to achieve 1 x 106 cells/50 µL. If the final volume of combined cells is greater than 50 µL, centrifuge at 4°C for 5 minutes at 400 – 600 x g. Following centrifugation, adjust the volume to 50 µL by removing the Cell Staining Buffer. After addition or removal of the Cell Staining Buffer, gently resuspend cells by pipetting.
- If staining cells with the TotalSeq-A Universal cocktail and the volume of combined cells is less than 25 µL, adjust volume with Cell Staining Buffer up to 25 µL to achieve 5 x 105 cells/25 µL. If the final volume of combined cells is greater than 25 µL, centrifuge at 4°C for 5 minutes at 400 – 600 x g. Following centrifugation, adjust the volume to 25 µL by removing the Cell Staining Buffer. After addition or removal of the Cell Staining Buffer, gently resuspend cells by pipetting.
- If you combined 6 samples at 10 µL each for a total of 60 µL and used an antibody pool or TotalSeq-A TBNK Cocktail, you would remove 10 µL after centrifugation for a total volume of 50 µL, resulting in 1 x 106 cells/50 µL.
- If you combined 4 samples at 10 µL each for a total of 40 µL and used a TotalSeq-A Universal Cocktail to stain cells, you would remove 15 µL of buffer after centrifugation for a total of 25 µL, resulting in 5 x 105 cells/25 µL.
- Proceed with staining your combined samples using either the Cell Staining with TotalSeq Antibody Pool section 5.2.1, or Cell Staining with TotalSeq-A Cocktail section 5.3.1 protocols below. Cells will already be stained with TotalSeq Hashtag Antibodies; do not add additional Hashtag Antibodies to the TotalSeq-A antigen-specific antibody pool.
- Proceed to step 5.2 if one of the following applies:
- Cell staining with TotalSeq Antibody Pool and TotalSeq Hashtags
- Prepare the antibody pool by combining titrated amounts of each specific TotalSeq-A antibody in a low protein binding microcentrifuge tube. For more information regarding titration of TotalSeq-A antibodies, please read Tips and Tricks for Titrating TotalSeq Antibodies. If you have additional questions, please reach out to BioLegend Technical Services.
- When performing dual staining with TotalSeq-A cell hashtag antibodies and TotalSeq-A antigen-specific antibodies, we recommend adding cell hashing antibodies into each respective sample’s TotalSeq-A antibody pool (figure 1A).
- If the antibody pool volume is less than 50 µL, adjust the volume with Cell Staining Buffer up to 50 µL. If the volume of the pool is above 50 µL, no volume adjustment is necessary.
- Centrifuge the antibody pool at 14,000 x g at 2 – 8°C for 10 minutes before adding to the cells. This is critical to ensure the removal of protein aggregates.
- Carefully pipette out the prepared antibody pool, avoiding the bottom of the tube, and add the TotalSeq antibody pool to the 50 µL blocked cell suspension.
- Incubate for 30 minutes at 4°C.
- Cell staining with TotalSeq-A Cocktail and TotalSeq Hashtags
- If staining cells with a the TotalSeq-A TBNK Cocktail or any other TotalSeq-A Cocktail, follow the reconstitution steps below.
- Equilibrate the lyophilized panel vial(s) to room temperature for 5 minutes.
- Place the lyophilized panel vial in an empty microcentrifuge tube, and spin down at 10,000 x g for 30 seconds at room temperature.
- If cell hashing samples simultaneously with a TotalSeq-A Cocktail, please refer to the TotalSeq Antibody Cocktail “Spike-in” Guidance protocol prior to reconstitution of the lyophilized cocktail to determine how to make the Cell Staining Buffer and spike-in reconstitution mix. Then proceed to the next step, 5.3.5, to reconstitute the lyophilized cocktail. If not hashing or if your samples are already stained with hashtags, proceed to the next step, 5.3.5.
- If using the TBNK cocktail, rehydrate by adding 50 µL of Cell Staining Buffer, or the Cell Staining Buffer and hashtag mix. If using any other TotalSeq-A Cocktail, rehydrate by adding 27.5 µL of Cell Staining Buffer or the Cell Staining Buffer and hashtag mix. Replace the cap and vortex for 10 seconds.
- Incubate at room temperature for 5 minutes.
- Vortex again and spin down at 10,000 x g for 30 seconds at room temperature.
- Transfer the entire volume (50 µL for the TotalSeq-A TBNK Cocktail or 27.5 µL for any other TotalSeq-A Cocktail) to a low protein binding microcentrifuge tube.
- Centrifuge at 14,000 x g for 10 min at 4°C. Important: Centrifugation of the reconstituted cocktail at 14,000 x g at 2 – 8°C for 10 minutes is critical to ensure the removal of antibody aggregates.
- Transfer 50 µL or 25 µL of the reconstituted cocktail you used to the tube containing 50 µL or 25 µL of FcR-blocked cells, taking care to avoid the bottom of the tube. The final staining volume will be 100 µL for cells stained with the TotalSeq-A TBNK Cocktail, or 50 µL for cells stained with any other TotalSeq-A Cocktail. Mix by gently pipetting 5 times.
- Incubate for 30 minutes at 4°C.
- If you plan to multiplex samples using TotalSeq hashtags, there are two commonly used approaches to staining samples with hashtag antibodies.
- Wash cells
- Add 3 mL of Cell Staining Buffer and mix by gently pipetting 5 times. Centrifuge at 4°C for 5 minutes at 400 – 600 x g depending on your sample type. Repeat this step twice for a total of 3 washes.
Note:
It is important to thoroughly decant the wash buffer and resuspend the cell pellet by either pipetting or gentle vortexing. Discard supernatant with a single firm, but not overly forceful motion. Proceed to absorb any remaining liquid on the lip of the tube with a clean paper towel.
- After the final wash, decant the supernatant and resuspend cells in 200 µL of Cell Staining Buffer for an approximate final volume of 250 – 350 µL. Gently mix the cells by pipetting.
- Slowly filter cells through 40 µm Flowmi™ Cell Strainer into a low protein binding microcentrifuge tube.
Note:
40 µm Flowmi™ Cell Strainer may be too small for some sample types.
- Count cells and assess cell viability.
- Adjust cell concentration using PBS according to the input requirements of your single-cell partitioning platform.
- Add 3 mL of Cell Staining Buffer and mix by gently pipetting 5 times. Centrifuge at 4°C for 5 minutes at 400 – 600 x g depending on your sample type. Repeat this step twice for a total of 3 washes.
II) Proceed with the Chromium Single Cell 3' Reagent Kits User Guide (v3.1- Dual Index) with Feature Barcoding technology for Cell Surface Protein, CG000317 as described through Post Gem-RT Cleanup – Dynabeads (step 2.1).
- At cDNA amplification step (Step 2.2), use the following table to prepare cDNA amplification mix:
ADT 1 rxn (µL) HTO 1 rxn (µL) ADT + HTO 1 rxn (µL) Amp Mix 50 50 50 cDNA Primers* 15 15 15 ADT Additive Primer (0.2 µM stock) 1 0 1 HTO Additive Primer v2 (0.2 uM stock) 0 1 1 Total 66 66 67 *included with 10x Genomics 3’ kit (PN 2000089), different from Feature cDNA primers 2.
- Add amplification mix to 35 µL of the sample.
- Mix by pipetting 15x with the pipette set to 90 µL. Centrifuge briefly.
- Proceed to the 10x User Guide CG000317 Rev B step 2.2D (thermal cycler protocol) of the cDNA Amplification.
Important: Follow steps 2.3A and 2.3B exactly to separate ADTs/HTOs from cDNA. Continue to use 70 µL of sparQ or SPRI beads in step 2.3B.
III) ADT and HTO Dual Index Library Preparation
Important:
- Use the DI_ADTx / DI_HTOx primer pairs specified in the primer table; do not mix primer pairs.
- For samples that contain both ADTs and HTOs, you will need to perform two separate reactions when preparing the dual index libraries, one for the ADT library and one for the HTO library. Use 5 µL of the “purified ADT/HTO fraction” from step 2.3B of the 10x Genomics protocol for each reaction and proceed with preparing the reaction mix using the table below.
- At Cell Surface Protein Library Construction (Step 4.1) in the 10x user guide CG000317, use the following table to prepare the Sample Index PCR Mix.
ADT HTO Volume (µL) Purified ADT/HTO fraction Purified ADT/HTO fraction 5 DI_ADTx i5 primer (10 µM stock) DI_HTOx i5 primer (10 µM stock) 2.5 DI_ADTx i7 primer (10 µM stock) DI_HTOx i7 primer (10 µM stock) 2.5 2X QuantaBio or Kapa Hifi Master Mix 2X QuantaBio or Kapa Hifi Master Mix 50 RNAse-free water RNAse-free water 40 Total 100 - Incubate in a thermal cycler with the following protocol:
ADT 98°C 2 min 98°C 20 sec 14 - 15 Cycles 60°C 30 sec 72°C 20 sec 72°C 5 min 4°C Hold HTO 98°C 2 min 98°C 20 sec 13 - 15 Cycles 64°C 30 sec 72°C 20 sec 72°C 5 min 4°C Hold - Perform post ADT/HTO library amplification clean-up.
- Add 120 µL sparQ or SPRIselect Reagent (1.2X) to each sample.
- Incubate 5 min at room temperature.
- Place on the magnet in its High position until the solution clears.
- Carefully remove and discard the supernatant.
- Place tubes on magnet in its High position. Wash the pellet twice with 200 µL 80% ethanol.
- Centrifuge briefly. Place on the magnet Low. Remove remaining ethanol.
- Remove from the magnet. Add 40.5 µL water.
- Incubate 2 min at room temperature.
- Place on the magnet in its Low position until the solution clears.
- Transfer 40 µL to a new tube strip. Store at 4°C for up to 72 h or at −20°C for long-term storage.
- ADT/HTO libraries are now ready to be sequenced. Quantify libraries by standard methods (QuBit, BioAnalyzer, qPCR). ADT and HTO libraries will be around 230 bp (figure 2).
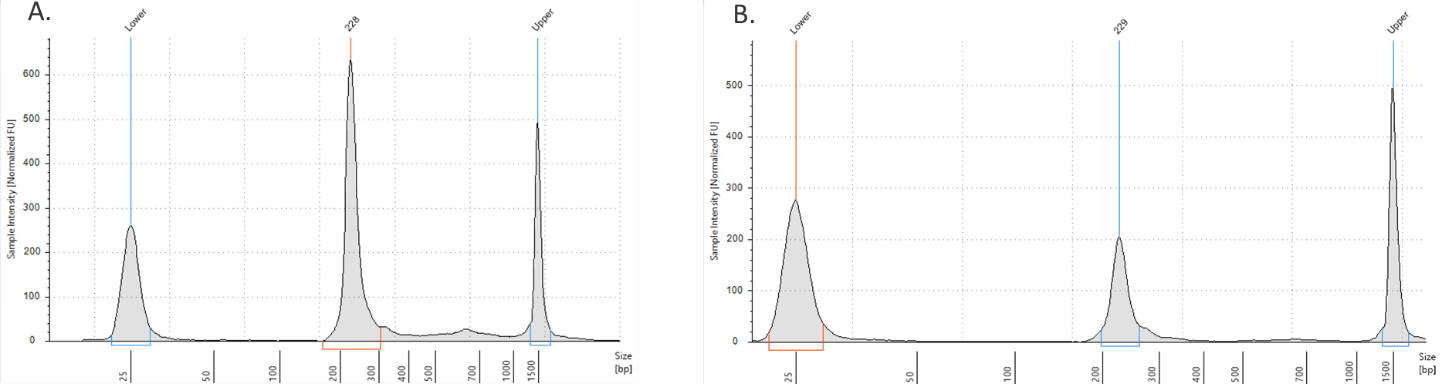
Figure 2. ADT/HTO library verification on the TapeStation D1000 tape. A) ADT Library product (~230 bp). B) HTO library product (~230 bp).
IV) Sequencing CITE-seq libraries:
To obtain sufficient read coverage for both libraries, we typically sequence ADT libraries in 10 - 20% of a lane and the gene expression library fraction at 80% - 90% of a lane (NovaSeq 6000 Flow Cell). This can vary depending on depth of coverage for each library you are trying to achieve. See table below for sequencing depth recommendations.
|
Library Type |
Minimum Sequencing Depth |
|---|---|
|
3' Gene Expression Library |
20,000 - 50,000 |
|
Cell Surface Protein Library <100 ADT panel |
5,000 |
|
Cell Surface Protein Library ≥100 ADT panel |
10,000 |
|
Cell Hashing Libraries |
500 |
TotalSeq™ A – Dual Index Read Length Requirements
Scenario 1. Sequencing Antibody (ADT and/or HTO) Library and Gene Expression GEX) Library together.
| Read1 | Index1 (i7) | Index2 (i5) | Read2 | |
| Number of cycles | 28 | 10 | 10 | 90 |
Scenario 2. Sequencing Antibody (ADT and/or HTO) Library only.
| Read1 | Index1 (i7) | Index2 (i5) | Read2 | |
| Number of cycles | 28 | 10 | 10 | 25 |
Notes regarding oligonucleotide sequences:
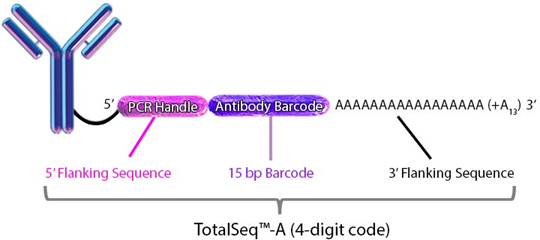
TotalSeq™ antibodies:
Each clone is barcoded with a unique oligonucleotide sequence. These contain standard small TruSeq RNA read 2 sequences and can be amplified using Dual Index ADT i5/i7 Primer Pairs (DI_ADTx) to obtain Illumina and 10x GEX dual-indexing compatible sequencing libraries): CCTTGGCACCCGAGAATTCCAAACAAGACCCTTGAGBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA*A*A.
Please visit our webpage for detailed information:
Oligos required for ADT and HTO library amplification:
- HPLC purified primers are preferred when ordering primers.
- The phosphorothioate bonds in the primer renders the oligonucleotide resistant to nuclease degradation.
- A unique Illumina index should be used for each 10x Genomics sample lane used within one sequencing lane.
cDNA Primers
- ADT cDNA PCR additive primer: 5’CCTTGGCACCCGAGAATT*C*C
- HTO cDNA PCR additive primer: v2 5’GTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T
Sequencing Library Primers
- Dual Index ADT i5/i7 Primer Pairs (DI_ADTx) (for ADT amplification; see table 1 for full sequences)
[DI_ADT1 i5] - 5’AATGATACGGCGACCACCGAGATCTACAC-[10 bp index]-ACACTCTTTCCCTACACGACGC*T*C
[DI_ADT1 i7] - 5’CAAGCAGAAGACGGCATACGAGAT-[10 bp index]-GTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A
- Dual Index HTO i5/i7 primer pairs (DI_HTOx) (for HTO amplification; see table 1 for full sequences)
(DI_HTO1) i5 - 5’AATGATACGGCGACCACCGAGATCTACAC-[10 bp index]-ACACTCTTTCCCTACACGACGC*T*C
(DI_HTO1) i7 - 5’CAAGCAGAAGACGGCATACGAGAT-[10 bp index]GTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T
* indicates a phosphorothioate bond.
B indicates C, G, or T nucleotide; not an adenine (A) nucleotide.
Table 1. Primers Used for Sequencing Library Construction
| Index | Full Sequence 5’ -> 3’ | SampleSheet Index1 |
SampleSheet Index2 (forward strand workflow) |
SampleSheet Index2 (reverse complement workflow) |
|
| DI_ADT1 | i5 | AATGATACGGCGACCACCGAGATCTACACCGTAGAGCAGACACTCTTTCCCTACACGACGC*T*C | TCTACTCGCG | CGTAGAGCAG | CTGCTCTACG |
| i7 | CAAGCAGAAGACGGCATACGAGATCGCGAGTAGAGTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A | ||||
| DI_ADT2 | i5 | AATGATACGGCGACCACCGAGATCTACACGACGAGATGCACACTCTTTCCCTACACGACGC*T*C | CTCTGCGCTA | GACGAGATGC | GCATCTCGTC |
| i7 | CAAGCAGAAGACGGCATACGAGATTAGCGCAGAGGTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A | ||||
| DI_ADT3 | i5 | AATGATACGGCGACCACCGAGATCTACACGATGGCGAACACACTCTTTCCCTACACGACGC*T*C | TCGCAGCTTC | GATGGCGAAC | GTTCGCCATC |
| i7 | CAAGCAGAAGACGGCATACGAGATGAAGCTGCGAGTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A | ||||
| DI_ADT4 | i5 | AATGATACGGCGACCACCGAGATCTACACCGAACGATGGACACTCTTTCCCTACACGACGC*T*C | CTAGTCGCCT | CGAACGATGG | CCATCGTTCG |
| i7 | CAAGCAGAAGACGGCATACGAGATAGGCGACTAGGTGACTGGAGTTCCTTGGCACCCGAGAATTC*C*A | ||||
| DI_HTO1 | i5 | AATGATACGGCGACCACCGAGATCTACACCGAAGGTCAGACACTCTTTCCCTACACGACGC*T*C | GCTTCACGCT | CGAAGGTCAG | CTGACCTTCG |
| i7 | CAAGCAGAAGACGGCATACGAGATAGCGTGAAGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T | ||||
| DI_HTO2 | i5 | AATGATACGGCGACCACCGAGATCTACACGAGTAACGGCACACTCTTTCCCTACACGACGC*T*C | CTCAGGTCTC | GAGTAACGGC | GCCGTTACTC |
| i7 | CAAGCAGAAGACGGCATACGAGATGAGACCTGAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T | ||||
| DI_HTO3 | i5 | AATGATACGGCGACCACCGAGATCTACACTGCAAGGACGACACTCTTTCCCTACACGACGC*T*C | TCTCTGCGAC | TGCAAGGACG | CGTCCTTGCA |
| i7 | CAAGCAGAAGACGGCATACGAGATGTCGCAGAGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T | ||||
| DI_HTO4 | i5 | AATGATACGGCGACCACCGAGATCTACACAATGGACGGCACACTCTTTCCCTACACGACGC*T*C | CTCGACGCTT | AATGGACGGC | GCCGTCCATT |
| i7 | CAAGCAGAAGACGGCATACGAGATAAGCGTCGAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGAT*C*T |
* Note: for more Dual Index ADT i5/i7 Primer Pairs (DI_ADTx) and Dual Index HTO (DI_HTOx) i5/i7 Primer Pairs, please refer to this table.
Representative Data and Troubleshooting
My ADT/HTO library contains a large peak at ~140-180 bp.
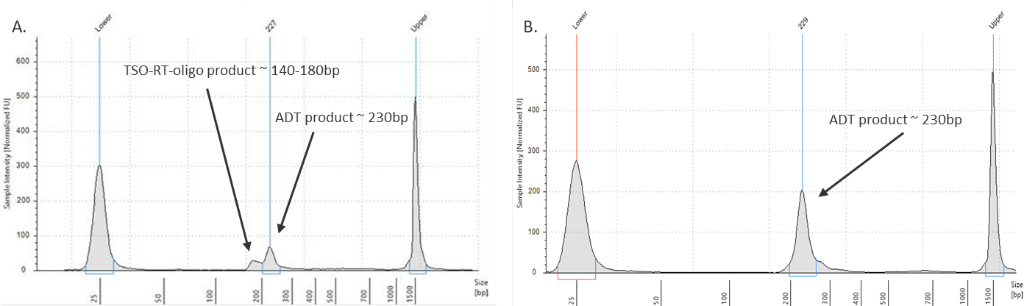
Figure 3. ADT library verification.
(Left graph) A TSO-RT-oligo product (~140 - 160 bp) can be amplified during the ADT PCR by carryover primers from cDNA amplification. The product will not cluster, but will interfere with quantification. Sequential 2X sparQ or SPRI purification of the ADT fraction after cDNA amplification reduces carryover of primers from cDNA amplification, and minimizes the amplification of this product during ADT library amplification. To further enrich for ADT specific product, the purified ADT library can be reamplified for three additional cycles with ADT specific primer sets or generic P5/P7 primers. A clean ADT library will contain a predominant single peak at around 230 bp (figure 3, right graph).
My ADT/HTO library contains a large peak at ~400 bp.
Overamplification of a library can lead to depletion of available primers and/or dNTPs resulting in self-priming of PCR products by their P5 and/or P7 adapters. This can lead to the production of “daisy-chains” or “bubble products”. These products consist of essentially two ADT or HTO barcode sequences attached to one another in one long oligo tag that is twice as long as the original oligo tag; these products appear as peaks at approximately 400 - 440 bp. These peaks can be more or less pronounced.
The larger peak is perfectly acceptable to sequence. However, it is difficult to quantify these libraries to titrate for sequencing. This error can be corrected by performing another PCR reaction using generic P5/P7 primers (not used in the protocol) for one or two cycles on the 1.2x sparQ or SPRI cleaned up product. The PCR reaction will convert the larger product to the desired size product partially or completely as the major peak.



Follow Us